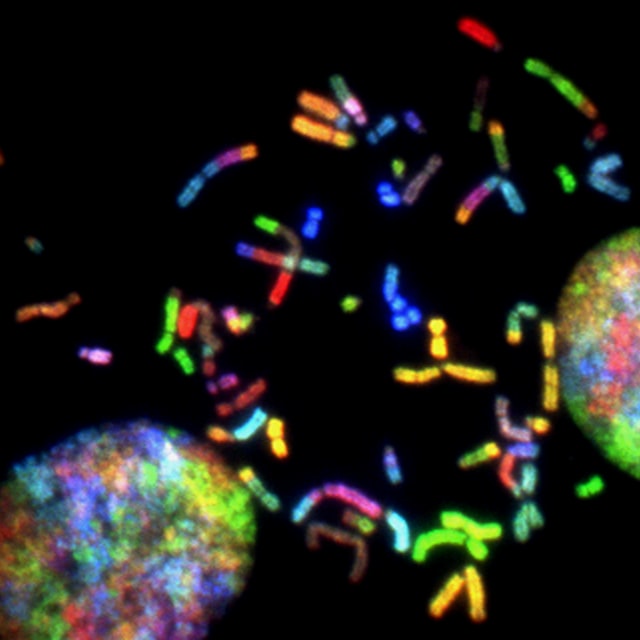




AIのEVE「遺伝子変異を良性か病気かを診断する」
人間の遺伝子変異を、病気か良性を診断するAIモデルが開発されました。
EVEと呼ばれるAIはDNA配列を隅々まで調べ判断します。
No two human beings are the same, a biologic singularity encoded in the unique arrangement of the molecules that make up our individual DNA.
参照元:https://hms.harvard.edu/news/all-about-eve
– ハーバード・メディカル・スクール Harvard Medical School. October 27, 2021 –
生物学的特異性は、DNAを構成する分子のユニークな配列に表れている。
変化は生物学の主要な特徴であり、多様性の原動力であり、進化の原動力でもあるが、その一方で暗黒面もあります。
DNAの配列が変化し、その結果として細胞を構成するタンパク質が変化すると、時として生理機能が大きく損なわれ、病気を引き起こすことがあります。
では、どのような遺伝子の変化が正常で、少なくとも取るに足らないもので、どのようなものが病気の前兆となるのでしょうか?
しかし、過去20年間にゲノム配列解析技術が飛躍的に進歩したにもかかわらず、それによって同定された何百万もの遺伝子変異の意味を解釈する能力は、まだ遅れています。
ハーバード大学医学部とオックスフォード大学の研究者たちは、EVE(Evolutionary model of Variant Effect)と呼ばれるAIツールを開発しました。
このツールは、高度な機械学習を利用して、人間以外の何十万もの種の遺伝子変異のパターンを検出し、それをもとに人間の遺伝子の変異の意味を予測します。
2021年10月27日にNature誌に掲載された解析結果では、EVEを用いて、複数の生物種にわたる3,600万のタンパク質配列と3,219の疾患関連遺伝子を評価しました。
その結果、これまでに同定された256,000個の意義不明のヒト遺伝子変異は、実際には良性または病気の原因のいずれかに分類し直されるべきであることが示唆されました。
このツールは、遺伝子変異の意味を判断するために現在用いられている臨床的手法を補強するために使用できるそうです。
また研究者たちは、「このようなツールと併用することで、診断、予後、治療法の選択の精度を高めることができるでしょう」と述べています。
今回の研究でオックスフォード大学のYarin Gal教授、ハーバード大学のJonathan Frazer教授とMafalda Dias教授、オックスフォード大学のPascal Notin教授と共同で、HMSのBlavatnik研究所でシステム生物学の准教授を務めるDebora Marks氏は話します。
「しかし、データの意味を理解することは必ずしも容易ではありません。「病気の可能性や病気の進行にどのような意味があるのか、ほとんど情報がありません。」
EVEは診断テストではありませんが、その計算能力は、遺伝学者やその他の医師が、特定の病気を引き起こす遺伝子変異の有無に基づいて診断を下し、病気の進行を予測し、さらには治療法を選択するために使用している現在の臨床ツールを強化することができると、研究者たちは強調しています。
Marks氏は話します。
「我々のアプローチは、現在の臨床評価の追加ツールとして使用でき、特に臨床現場において、不確実性を低減し、意思決定を明確にする強力な新しい方法を提供できると考えています。」
実際、EVEは、臨床効果の予測において、他の計算機上の予測モデルよりも優れており、また、変異が生物学的機能に及ぼす影響を検証する現在のゴールドスタンダードのハイスループット実験と同等以上のスコアを示しました。
遺伝的変異の意味を正確に解釈することは、非常に大きな意味を持ちます。
良性の変異を病気の原因とみなすと、誤った診断が下され、さらなる検査や不安、さらには不必要な医療介入が連鎖的に行われることになります。
逆に、自分のDNA上の病気の原因となる変化を、取るに足らない良性のものと誤認してしまうと、注意深い観察やさらなる検査、予防措置が必要な場合に、誤った安心感を与えてしまうことになります。
Gal氏は話します。
「私たちが期待しているのは、現場の臨床医が正しい診断、予後、治療を行うための強力なデータを得ることです。」
データが増えれば疑問も増える
2003年に行われた歴史的なヒトゲノムの解読により、新たに解読されたゲノムと比較するための基準となるヒトゲノムが確立されました。
しかし、この基準ゲノムは、「正常な」ヒトゲノムの標準や基準値ではありません。
急速に増加しているDNA配列のデータは、基準ゲノムを標準というよりも、研究者が遺伝的変異の意味をよりよく理解するのに伴って時間とともに変化する流動的な基準にしています。
ヒトゲノムの特定の変化と疾患の発生を関連付けることは、臨床遺伝学の分野では依然として困難な課題です。
なぜなら、ヒト集団における変異の数は、科学者が調査できる数よりも多いからです。
ヒト集団のごく一部しか配列が決定されていないにもかかわらず、研究者たちはすでに、意味や意義がはっきりしない何百万ものバリアントを目にしています。
それらの変異体のうち、良性、中性、病原性のいずれかに分類されるものはわずか2%です。
残りの98%の遺伝子変異は、現在のところ「意義不明」とされています。
ヒトゲノムでは、タンパク質をコードする領域だけでも、遺伝子によって作られるタンパク質の中の1つのアミノ酸の位置に関わる650万個の変異が数百万個確認されています。
これらのミスセンス変異は、タンパク質の機能に影響を与えない場合もあれば、タンパク質の機能を低下させて病気を引き起こす場合もあります。
実際、研究者たちは、地球上に住む90億人の人々のゲノムの中には、致死的なものを除いて、すべてのタンパク質の位置に変異があるのではないかと推測しています。
一人ひとりのゲノムには、他の人や基準となるヒトゲノムと比較して、多くのバリアントが存在します。
さらに、人間はそれぞれの親から2種類の遺伝子を受け継いでいます。
また、加齢に伴い、体細胞変異と呼ばれる遺伝子の変化が起こることもあります。
Marks氏は話します。
「1人の人間が1つのゲノムだけを持っているわけではなく、さまざまな方法があります。また、加齢に伴い、あらゆる種類の体細胞変異が発生します。これは、がんの発生に関連するだけでなく、神経変性にも関連しており、いずれも変異によって引き起こされる加齢関連プロセスです。」
確かに、乳がんや卵巣がんのBRCA1やBRCA2、さまざまながんの腫瘍抑制遺伝子p53など、研究者が臨床疾患のリスクが高い変異を特定した疾患関連遺伝子は数多くあります。
しかし、それらの遺伝子でさえ、他の研究されていない突然変異が見られ、その意義はいまだに不明です。
これらのことから、人間の遺伝子変異の意義を明らかにすることが急務であり、その答えを出すために計算機がますます重要な役割を果たすことになるだろうとMarks氏は述べています。
AIの登場
ニューラルネットワークの特徴は、新しいデータが得られたときに、仮説の確率を継続的に再評価し、更新する能力を持っていることです。
つまり、ニューラルネットワークは、新しい知識を用いて証拠を再評価し、従来の方法では見逃していたパターンや意味を検出することができるのです。
今回の研究では、「教師なし機械学習」と呼ばれる高度な分析手法を用いました。
これは、あらかじめ定義されたパラメータやルールに基づくのではなく、適応的な学習を行う人工知能の一形態です。
つまり、新しいデータを受け取った機械学習アルゴリズムは、時間の経過とともにパターンの認識がうまくできるようになるということです。
一方、教師付き機械学習では、アルゴリズムはあらかじめラベル付けされたデータからパターンを検出することを学習します。
情報技術者の典型的な例では、アルゴリズムは、猫と犬の画像を提示され、どれがどれであるかを教えられた後、ラベルのない猫と犬の画像を認識するように指示されます。
教師なし機械学習では、アルゴリズムは猫と犬の画像のセットを与えられ、どれがどれだかは教えられません。自分自身でパターンを識別する必要があります。
どちらのタイプも、特定のタスクに適しています。
教師なしモデルの利点は、事前にラベル付けされたデータを与えることで、学習に偏りが生じる可能性がないことです。
また、データの変化に対応して、より複雑な分析を行うことができます。
現在、遺伝子変異の重要性を評価するために用いられているほとんどの計算手法は、臨床的なラベルに基づいた教師付き学習を採用していますが、これではツールに偏りが生じ、現実世界での予測精度が高くなってしまう可能性があります。
Gal氏は話します。
「アルゴリズムは、どの画像が猫で、どの画像が犬なのかを事前に知る必要はなく、猫と犬の画像がたくさんあればよいので、知らないはずの情報を利用することはできません。」
教師なし機械学習は、これまでに出会ったことのないデータから新たなパターンを検出する能力があるため、人間以外の遺伝子配列の解析に適しているのです。
進化した親戚からのヒント
今回の研究では、複数の種の遺伝子変異を調べることで、ヒトの変異の重要性を知る手がかりが得られるのではないかという、古くからの期待が寄せられている。
進化は、機能や生存に不可欠な、あるいは少なくとも重要な特徴を、種を超えて保存する傾向がある。
したがって、種を超えて繰り返されるアミノ酸配列は、生物学的に重要なマーカーであり、生物の機能や進化的な適合性にとって重要であることを示している。
したがって、このような高度に保存された配列に変化が生じると、問題が発生する可能性が高くなり、病原性につながると考えられます。
Marks氏は話します。
「これらの種は、進化的にはかなり離れていますし、遺伝子的にも多くの違いがありますが、それらをまとめて考えることで、私たちに情報を与えてくれます。このモデルが、人間や人間のバリエーションに関連するパターンについて、非常に強力な理由です。」
EVEは、結論を導くために、進化的に保存されたパターンを探しました。
EVEでは、絶滅危惧種を含む14万種のデータを分析しましたが、その中には絶滅した生物も含まれていました。
科学者たちは長年にわたり、比較遺伝学を用いて、DNAやタンパク質の配列間の類似性のある領域を検出し、意味を導き出してきました。
ハーバード・オックスフォード大学のチームは、ニューラルネットワークを使って、より大きなスケールでそれを実現しました。
EVEの学習
EVEは、2億5,000万個のタンパク質配列を用いて学習した後、1つ1つのアミノ酸バリアントが良性か病原性かの可能性を推定しました。
EVEが正確な予測を行っているかどうかを判断するために、研究チームはEVEのスコアを、重要性が知られているヒトの既存の変異と比較しました。
その結果、EVEの予測結果は、臨床データと非常によく一致していることがわかりました。
次に、病気に関連する3,219個のヒト遺伝子にEVEを適用しました。
EVEは、60個の「臨床的に対処可能な」遺伝子を含むすべての遺伝子について、その変異が病原性か良性かを正しく判断したそうです。
EVEの性能を、他の教師あり、教師なしのツールと比較したところ、格段に高い予測精度を示しました。
しかし、遺伝子の変異が生理機能にどのような影響を与えるかを評価するためのゴールドスタンダードである、実際の臨床実験で得られた知見と比較した場合、EVEの予測はどのようになるのでしょうか。
そこで研究チームは、さまざまな種類のがん、いくつかのがん症候群、心調律障害に関連する遺伝子のうち、よく研究されている5つの遺伝子の変異を対象とした臨床実験の結果とEVEのスコアを比較しました。EVEの予測は、実験データから得られた現在のラベルと重なりました。
Marks氏は話します。
「私たちの予想をはるかに上回る結果が得られました。進化に伴う配列の分布に合わせてモデルを学習させるだけで、特定の遺伝子変異から生じる疾患リスクについて予想外に正確な予測を可能にする情報が得られたようです。」
信頼の問題
EVEが現在の手法に比べて優れている点は、バイナリスコアではなく連続スコアを割り当てることです。
これは、遺伝子変異が良性か病原性かのラベルを付けられていても、その変異が生理学的にどのように現れるかは、より微妙なものだからです。
Marks氏は話します。
「病原性には連続性があります。連続スコアは、病原性のレベルを予測するのに非常に重要です。突然変異によって足の小指が痛くなるのか、それとも明日死んでしまうのか。」
このツールのもう一つの重要な点は、遺伝子ごとに予測の信頼度を示すスコアが割り当てられていることです。
これは、臨床医が予測の確実性の度合いを把握するのに役立ちます。
言い換えれば、遺伝子変異ごとに、EVEは専門家にその判断をどの程度信頼できるかを伝えます。
これは信頼性の問題であり、モデルに対する自信の問題であると研究者は述べています。
Gal氏は話します。
「私たちは臨床家に単に数値を提供するのではなく、その数値に付随する不確実性の度合いも提供しています。これは、専門家が、意思決定の過程で利用できるものです。このツールは、『あのバリアントはあの山に属していると思うが、これまでにそのようなバリアントを見たことがないので、大目に見てほしい』と言うことができます。」
「また、ツールは「その別のバリアントはこのパイルに属すると思うが、過去にそれと非常に似たバリアントを見たことがあり、それらがこのパイルに属すると見たので、自信を持ってこのパイルに割り当てよう」と言うこともできます。ツールと専門家の間に信頼関係を築くことは、この仕事の重要な側面です。」
今後の展望
研究者らは、この種のモデリングはまだ初期段階にあり、進化と遺伝子変異が病気についてまだまだ多くのことを教えてくれることは明らかであると述べ、タンパク質コード領域以外のゲノムの他の部分にも研究を拡大する予定であると付け加えました。
しかし、当面の課題は、ある程度の理解が得られている遺伝的変異を臨床的に利用することである。
そのために、研究者たちはすでにゲノム配列解析の会社と提携し、Chan Zuckerberg Initiativeを通じてさまざまなグループと協力しています。
また、「Atlas of Variant Effects Alliance」にも参加しています。
これは、ゲノム上の変異の影響をマッピングし、ヒトの遺伝子変異の可能性と、それがタンパク質の機能や生理に及ぼす影響を網羅したアトラスを作成することを目的とした、世界的な研究活動です。
このプロジェクトの最終的な目標は、ヒトの病気の診断、予後、治療を改善することです。
本研究の共著者には、オックスフォード大学のエイダン・ゴメス氏、ハーバード・メディカル・スクールのジョセフ・ミン氏とケリー・ブロック氏が含まれています。
本研究は、Chan Zuckerberg Initiative(CZI2018-191853)、米国国立衛生研究所(R01GM120574)、GSKおよび英国工学・物理科学研究評議会(EPSRC ICASE award No.18000077)から一部の支援を受けています。

関連記事






新着記事


よく読まれている記事
-
 なぜタイピングより手書きの方が、記憶に定着するのか
なぜタイピングより手書きの方が、記憶に定着するのか -
どんな曲が好き?「 音楽の好みと性格の関連性は普遍的 」
-
視覚と意思決定領域の結びつきが強い「鮮明なイメージ能力がある人」
-
「触覚が敏感な部位はなぜあるのか」触覚メカニズムが解明される
-
不活性化されたリチウムイオン電池を甦えさせる「復活するリチウムイオン電池」
-
記憶が脳に保存される新しい理論「MeshCODE理論」が開発される
-
大面積有機フォトダイオードに置き換わる?「シリコンフォトダイオード技術」
-
幸福度を7%上げる「旅行の仕方」
-
「世界最長寿記録を更新し132歳まで生きる人が出現する」ベイズ統計学予測
-
組織の中で行われたインシビリティ(非礼な言動)を軽視してはいけない理由









N E W S & P O P U L A R最 新 記 事 & 人 気 記 事
WHAT'S NEW !!
-


男女ともに長生きになる「男女平等」
【男女ともに長生きになる「男女平等」】 権利とは人間が作り出した構造ですが、男女平等が進むと男女ともに長生きになるようです。 The first global study to investi... -

他者を犠牲にして利益を取る・利益を度外視して他者への害を取り除く
【他者を犠牲にして利益を取る・利益を度外視して他者への害を取り除く】 他者を犠牲にして自分の利益を選ぶ、自分にとって利益は少ないが他者への害を防ぐ、道徳的なに... -

「寿命を延ばす」良質な睡眠
【「寿命を延ばす」良質な睡眠】 良質な睡眠をとることは、寿命を何年も長くする可能性があります。 Getting good sleep can play a role in supporting your heart and... -

見極める力を養う「チャットボットの精度」
【見極める力を養う「チャットボットの精度」】 ChatGPTをはじめ、チャットボットの精度は人が書いたものかどうかわからない程までの水準になっています。 The most rec...
News
- 新着記事 -
Popular
- 人気記事 -
H A P P I N E S S幸 福
人気 (❁´ω`❁)




























M E A L食 事


























B R A I N脳
人気 (❁´ω`❁)


























H E A L T H健 康




















人気 (❁´ω`❁)
-

人体・脳
健康な脳を保ち老化を遅らせる「アマゾンの先住民族ツィマネ族の生活習慣」
【健康な脳を保ち老化を遅らせる「アマゾンの先住民族ツィマネ族の生活習慣」】 ボリビア・アマゾンの先住民族であるツィマネ族が、アメリカやヨーロッパの人々に比べて... -

社会
自制心が健康と若さをもたらす理由
【自制心が健康と若さをもたらす理由】 デューク大学の研究チームは、自制心が心身に及ぼす影響を調査しました。 1000人を出生から45年間に渡って追跡した大規模調査で... -

健康
高強度インターバルトレーニングは、適度な運動よりも心臓を強化する
【心臓を強化する高強度インターバルトレーニング】 ノルウェー科学技術大学の研究によると、トレーニングの強度が、病気の重症度を軽減し、心臓機能を改善し、作業能力...
-
人体・脳
健康な脳を保ち老化を遅らせる「アマゾンの先住民族ツィマネ族の生活習慣」
【健康な脳を保ち老化を遅らせる「アマゾンの先住民族ツィマネ族の生活習慣」】 ボリビア・アマゾンの先住民族であるツィマネ族が、アメリカやヨーロッパの人々に比べて... -
社会
自制心が健康と若さをもたらす理由
【自制心が健康と若さをもたらす理由】 デューク大学の研究チームは、自制心が心身に及ぼす影響を調査しました。 1000人を出生から45年間に渡って追跡した大規模調査で... -
健康
高強度インターバルトレーニングは、適度な運動よりも心臓を強化する
【心臓を強化する高強度インターバルトレーニング】 ノルウェー科学技術大学の研究によると、トレーニングの強度が、病気の重症度を軽減し、心臓機能を改善し、作業能力...
J O B仕 事
人気 (❁´ω`❁)














































T E C H N O L O G Y技 術

















人気 (❁´ω`❁)




















